Priones: qué son, características, estructura, funciones, enfermedades
¿Qué son los priones?
Los priones son proteínas sin genoma ni ácidos nucleicos que actúan como agentes infecciosos. Se encuentran en la membrana de células normales, solo que como proteínas mal plegadas y/o con estructura tridimensional anormal.
Estas proteínas son las responsables de múltiples enfermedades degenerativas y de muy alta mortalidad que afectan los tejidos neurales y la estructura del cerebro.
Son también llamadas enfermedades priónicas. Entre las más importantes que afectan a humanos están el kuru, la enfermedad de Gerstmann-Sträussler-Scheinker, el síndrome de Creutzfeldt-Jakob y el insomnio familiar fatal.
Características de los priones
– Los priones son estructuras proteicas presentes en las membranas celulares. Estas proteínas poseen una forma o una conformación alterada [PrP (Sc)].
– Con respecto a su multiplicación, se logra mediante la conversión de formas, como en el caso de la enfermedad de la tembladera. En esta enfermedad, los priones reclutan a las PrP (C) (las proteínas priónicas de conformación no alterada) para estimular la conversión hacia la isoforma PrP (Sc).
– Estas inusuales proteínas capaces de propagarse, no presentan ácidos nucleicos. Prueba de ello es que son resistentes a los rayos X y a la radiación ultravioleta. Estos agentes fácilmente descomponen los ácidos nucleicos.
– Las proteínas priónicas, de las que están compuestos los priones (PrP), se encuentran en todo el cuerpo, no solo de los seres humanos sino de otros vertebrados sanos.
– Algunos investigadores han logrado demostrar que, en ratones, estas proteínas activan la reparación mielínica en las células del sistema nervioso periférico. También ha sido demostrado que la ausencia de estas causa la desmielinización de tales células nerviosas.
Estructura de los priones
El conocimiento que se tiene sobre la estructura de los priones reside principalmente en las investigaciones realizadas en la bacteria Escherichia coli.
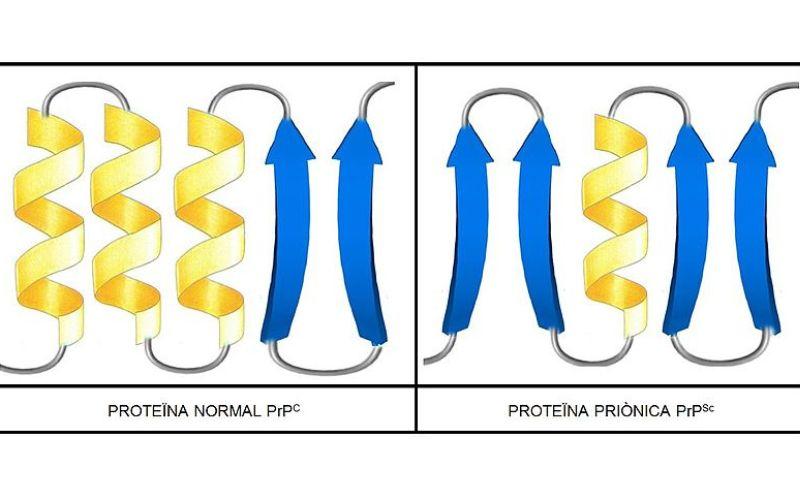
Los estudios hechos demuestran que los polipéptidos en cadena PrP (C) (normal) y PrP (Sc) (infeccioso) son idénticos en la composición de aminoácidos, pero difieren en la conformación 3D y en el plegamiento de estas.
PrP (C)
Estos priones no infecciosos presentan, en los seres humanos, 209 aminoácidos. Tienen un enlace disulfuro. Su estructura es alfa-helicoidal, lo que significa que tiene aminoácidos en forma de espiral (hélices alfa) y pocas hebras planas de aminoácidos (hojas beta).
Esta proteína no puede ser separada por centrifugación, lo que implica que no es sedimentable. Es fácilmente digerida por la serina proteasa de amplio espectro denominada proteinasa K.
PrP (Sc)
Es una proteína infecciosa que transforma PrP (C) en isoformas PrP (Sc) infecciosas y con una configuración o forma anormal.
Se sabe muy poco de su estructura 3D, sin embargo, se conoce que tiene pocas formas helicoidales y más hebras planas u hojas betas. El cambio hacia la isoforma es lo que se conoce como el evento fundamental de las enfermedades priónicas.
Funciones de los priones
Las proteínas priónicas celulares [Prp (C)] se ubican en la superficie celular de una gran variedad de órganos y tejidos. Se conoce muy poco acerca de las funciones fisiológicas de los priones en el organismo.
Aun así, experiencias hechas en ratones indican posibles funciones, tales como:
Con receptores metabotrópicos de glutamato
Ha sido demostrado que los PrP (C) actúan con los receptores de glutamato (los ionotrópicos y los metabotrópicos). El PrP (C) participa como un receptor de los oligómeros sinaptotóxicos del péptido Aβ de la superficie celular.
En el desarrollo embrionario
En ratones de la familia Murinae, se ha descubierto que las proteínas priónicas PrP (C) se expresan a los pocos días después de la implantación, en el desarrollo embrionario.
Esto indica que juegan un papel durante el desarrollo de estos pequeños mamíferos. Papel que según los investigadores está relacionado con la regulación de la neuritogénesis (producción de axones y dendritas de las neuronas).
También actúan en el crecimiento axonal. Estas proteínas priónicas incluso están involucradas en el desarrollo del circuito cerebeloso. Debido a ello, se cree que la ausencia de estos priones PrP (C) conlleva un retraso en el desarrollo motor de los roedores.
Neuroprotector
En estudios sobre la sobreexpresión de PrP (C) por la orientación de genes, se encontró que la ausencia de estos priones origina problemas con la irrigación sanguínea hacia algunos lugares del cerebro (isquemia cerebral aguda).
Esto significa que las proteínas priónicas funcionan como neuroprotectores. Adicionalmente, ha sido demostrado que la sobreexpresión de PrP (C) puede reducir o mejorar lesiones causadas por isquemia.
Sistema nervioso periférico
Recientemente, fue descubierta la función fisiológica de los Prp (C) en el mantenimiento de la mielina periférica.
Durante un estudio de laboratorio se descubrió que en ausencia de la proteína priónica, los ratones de laboratorio desarrollaron deficiencias de nervios que trasladan la información desde el cerebro y la médula espinal, en lo que se llama una neuropatía periférica.
Muerte celular
Existen algunas proteínas similares a los priones, y se localizan en otras partes del cuerpo distintas al cerebro.
Las funciones de tales proteínas es la de iniciar, regular y/o controlar la muerte celular, cuando el organismo está siendo atacado (por virones, por ejemplo), previniendo así la propagación del patógeno.
Esta función peculiar de estas proteínas, hace pensar a los investigadores sobre la posible importancia de los priones no infecciosos en la lucha contra patógenos.
Memoria a largo plazo
Un estudio realizado en el Instituto Stowers, en Missouri, EE. UU., demostró que los priones PrP pueden tener una función en el mantenimiento de la memoria a largo plazo.
El estudio reveló que ciertas proteínas priónicas pueden ser controladas para que trabajen en el mantenimiento de las funciones fisiológicas de la memoria a largo plazo.
Renovación de células madres
Una investigación sobre proteínas priónicas que se expresan en células madres del tejido sanguíneo, reveló que todas estas células madres (hematopoyéticas), expresan proteínas priónicas en su membrana celular. Por lo que se cree que participan en el complejo e importantísimo proceso de la renovación celular.
Enfermedades causadas por priones
Las enfermedades priónicas más comunes son:
Enfermedad de Creutzfeldt-Jakob (ECJ)
Considerada la enfermedad priónica más común entre los seres humanos, es una patología cosmopolita, es decir, de distribución mundial. Puede presentarse de forma hereditaria (familiar), esporádica o infecciosa.
Enfermedad de Gerstmann-Sträussler-Scheinker
Es una enfermedad causada por priones en un proceso encefálico infeccioso heredable o autosómico dominante. La enfermedad se manifiesta en personas de 40 a 60 años.
Prionopatía con sensibilidad variable a la proteasa
Es una enfermedad muy poco frecuente, al punto de que su rango de ocurrencia es de 2 a 3 casos por cada 100 millones de habitantes. La patología es similar a la enfermedad Gerstmann-Sträussler-Scheinker.
Insomnio letal
Es una enfermedad hereditaria o familiar, aunque también puede presentarse de forma esporádica. Se conoce que la enfermedad se debe a una mutación hereditaria o autosómica dominante.
Kuru
Esta enfermedad priónica ha sido detectada únicamente en habitantes de Papúa Nueva Guinea. Es una enfermedad relacionada con el canibalismo y la tradición cultural del rito del duelo por los muertos, en donde estas personas comen el encéfalo humano.
Enfermedades en animales
Entre las patologías producidas por priones en animales está la encefalopatía espongiforme bovina. Esta enfermedad causó estragos en Europa, en la salud pública, la de los animales y en la economía de los países afectados.
Otras enfermedades en animales son el scrapy, la encefalopatía transmisible del visón, la enfermedad del desgaste crónico (en ciervos) y la encefalopatía espongiforme felina.
Estas enfermedades, al igual que las presentadas en humanos, carecen de un tratamiento efectivo, por lo que la prevención es fundamental, sobre todo después de los contagios en humanos ocurridos a raíz del consumo de carne de vacas infectadas.
Tratamientos
Hasta la fecha no se conoce cura para las enfermedades priónicas. El tratamiento es sintomático. Se les aconseja a los pacientes planificar cuidados paliativos y se les recomienda análisis y asesoramiento genético para familiares.
Ha sido probada una gran variedad de medicamentos en pacientes con enfermedades priónicas, como antivirales, antitumorales, fármacos para enfermedades como el Parkinson, tratamientos para la inmunosupresión, antibióticos, antifúngicos, incluso antidepresivos.
Sin embargo, no existe evidencia en la actualidad que indique que algunos de estos disminuyan los síntomas o mejoren la supervivencia de los enfermos.
Prevención
Los priones son resistentes a una variedad de cambios físicos y químicos. Sin embargo, diferentes técnicas se emplean para evitar la contaminación de pacientes con instrumentos quirúrgicos contaminados.
Entre las técnicas más utilizadas está la de esterilizar los equipos en una autoclave a 132 °C por una hora y luego sumergir los instrumentos en hidróxido de sodio por al menos una hora más.
Por otra parte, la Organización Mundial de la Salud (OMS), ha desarrollado medidas para evitar el contagio de las enfermedades priónicas. Esta organización establece normas para el manejo de tejidos prohibidos o potencialmente riesgosos como: ojos, cerebro, intestino, amígdalas y médula espinal.
Referencias
- Prion, Infectious Agent. Recuperado de britannica.com.
- ¿What is a Prion? Recuperado de scientificamerican.com.
- Prion. Recuperado de en.wikipedia.org