Succinato deshidrogenasa: estructura, función, regulación, enfermedades
Succinato deshidrogenasa (SDH), también conocido como el complejo II de la cadena transportadora de electrones, es un complejo proteico mitocondrial con actividad enzimática que funciona tanto en ciclo de Krebs como en la cadena transportadora de electrones (respiración celular).
Es una enzima que está presente en todas las células aeróbicas. En los eucariotas se trata de un complejo estrechamente asociado con la membrana mitocondrial interna, mientras que en los procariotas se encuentra en la membrana plasmática.
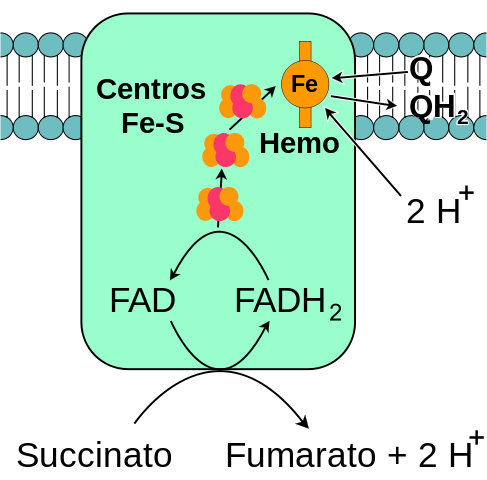
El complejo succinato deshidrogenasa, descubierto alrededor de 1910 y purificado por primera vez en 1954 por Singer y Kearney, ha sido extensamente estudiado por varios motivos:
– funciona tanto en el ciclo de Krebs (ciclo del ácido cítrico o ciclo de los ácidos tricarboxílicos) como en la cadena transportadora de electrones (cataliza la oxidación de succinato a fumarato)
– su actividad es regulada por distintos activadores e inhibidores y
– es un complejo asociado con: hierro no ligado a un grupo hemo, azufre lábil y dinucleótidos de flavina adenina (FAD)
Es codificado por el genoma nuclear y se ha comprobado que mutaciones en los cuatro genes que codifican cada una de sus subunidades (A, B, C y D) resultan en diversos cuadros clínicos, es decir, pueden ser bastante negativas desde el punto de vista de la integridad física de los seres humanos.
Índice del artículo
- 1 Estructura
- 2 Función
- 3 ¿Cómo actúa?
- 4 Regulación
- 5 Deficiencia de succinato deshidrogenasa
- 6 Enfermedades relacionadas
- 7 Referencias
El complejo enzimático succinato deshidrogenasa está formado por cuatro subunidades (heterotetrámero) codificadas por el genoma nuclear, por lo que es el único complejo de fosforilación oxidativa en la cadena transportadora de electrones que no posee ninguna subunidad codificada por el genoma mitocondrial.
Además, este complejo es el único que no bombea protones a través de la membrana mitocondrial interna durante su acción catalítica.
De acuerdo con estudios realizados en base al complejo enzimático de células cardíacas porcinas, el complejo succinato deshidrogenasa consiste en:
– una “cabeza” hidrofílica que se extiende desde la membrana mitocondrial interna hacia la matriz mitocondrial y
– una “cola” hidrofóbica que está embebida en la membrana mitocondrial interna y que posee un pequeño segmento que se proyecta hacia el espacio intermembrana soluble de la mitocondria

La cabeza hidrofílica está compuesta por las subunidades SdhA (70 kDa) y SdhB (27 kDa) (Sdh1 y Sdh2 en levaduras) y esta comprende el centro catalítico del complejo.
Las subunidades SdhA y SdhB contienen los cofactores redox que participan en la transferencia de electrones hacia la ubiquinona (coenzima Q10, molécula que transporta electrones entre los complejos respiratorios I, II y III).
La subunidad SdhA posee un cofactor FAD (una coenzima que participa en reacciones de óxido-reducción) unido covalentemente a su estructura, justo en el sitio de unión para el succinato (el principal sustrato de la enzima).
La subunidad SdhB tiene 3 centros hierro-azufre (Fe-S) que median la transferencia de electrones hacia la ubiquinona. Uno de los centros, 2Fe-2S, está cerca del sitio FAD de la subunidad SdhA y los otros (4Fe-4S y 3Fe-4S) son adyacentes al primero.
Cabe destacar que los estudios estructurales indican que la subunidad SdhB forma la interfase entre el dominio catalítico hidrofílico y el dominio de “ancla” a la membrana (hidrofóbico) del complejo.
El dominio membranal del complejo, como se dijo, consiste en las subunidades SdhC (15 kDa) y SdhD (12-13 kDa) (Sdh3 y Sdh4 en levaduras), que son proteínas integrales de membrana formadas, cada una, por 3 hélices transmembranales.
Este dominio contiene una porción hemo b unida en la interfase entre las subunidades SdhC y SdhD, donde cada una provee uno de los dos ligandos de histidina que las mantienen unidas.
En esta enzima se han detectado dos sitios de unión para ubiquinona: uno de gran afinidad y otro de baja afinidad.
El sitio de alta afinidad, conocido como Qp (p por proximal) se encuentra de cara hacia la matriz mitocondrial y está formado por residuos de aminoácidos específicos ubicados en las subunidades SdhB, SdhC y SdhD.
El sitio de baja afinidad, también denominado Qd (d por distal) está, en la porción de la membrana mitocondrial interna donde se inserta el complejo, más cerca del espacio intermembrana, es decir, más lejos de la matriz del orgánulo.
En conjunto, el complejo total tiene un peso molecular cercano a los 200 kDa y se ha determinado que tiene una relación de 4.2-5.0 nanomoles de flavina por cada miligramo de proteína y de 2-4 g de hierro por cada mol de flavina.
El complejo enzimático succinato deshidrogenasa cumple una importante función en la mitocondria, pues no solo participa en el ciclo de Krebs (donde participa en la degradación del acetil-CoA), sino que también forma parte de la cadena respiratoria, fundamental para la producción de energía en forma de ATP.
En otras palabras, es una enzima clave para el metabolismo intermediario y la producción aeróbica de ATP.
– Se encarga de la oxidación del succinato a fumarato en el ciclo del ácido cítrico
– Alimenta el complejo III de la cadena transportadora de electrones con los electrones derivados de la oxidación del succinato, lo que ayuda a reducir oxígeno y formar agua
– El transporte de electrones genera un gradiente electroquímico a través de la membrana mitocondrial interna, lo que favorece la síntesis de ATP
Como alternativa, los electrones pueden ser empleados para reducir moléculas de un “pool” de ubiquinonas, produciendo los equivalentes reductores necesarios para reducir aniones superóxido que se originan de la misma cadena respiratoria o que provienen de fuentes exógenas.
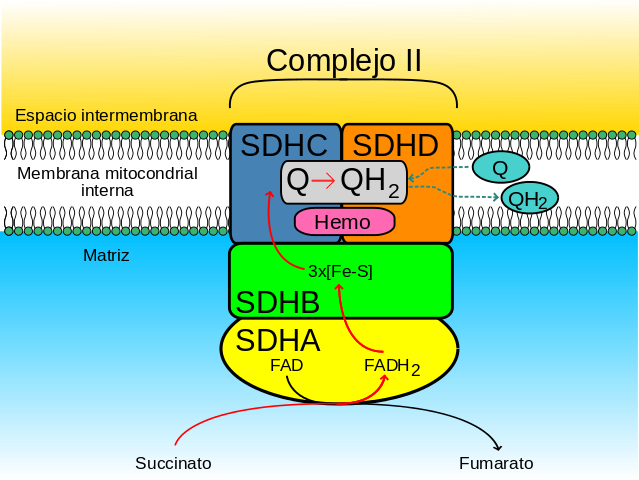
La subunidad A del complejo (la que está unida covalentemente a la coenzima FAD) se une a los sustratos, fumarato y succinato, así como a sus reguladores fisiológicos, el oxalacetato (inhibidor competitivo) y el ATP.
El ATP desplaza la unión entre el oxalacetato y el complejo SDH y, entonces, los electrones que son “pasados” desde el succinato hacia la subunidad SdhA son transferidos hacia los grupos de átomos de hierro y azufre presentes en la subunidad SdhB por medio de la coenzima FAD.
Desde la subunidad B, dichos electrones alcanzan los sitios hemo b de las subunidades SdhC y SdhD, desde donde son “entregados” a coenzimas quinonas a través de sus sitios de unión a quinonas.
El flujo electrónico desde el succinato a través de estos transportadores y hasta el aceptor final, que es el oxígeno, está acoplado a la síntesis de 1.5 moléculas de ATP por cada par electrónico a través de la fosforilación ligada a la cadena respiratoria.
Se ha reportado que mutaciones en el gen que codifica para la subunidad A del complejo succinato deshidrogenasa pueden causar encefalopatías durante la infancia, entretanto las mutaciones en los genes que codifican las subunidades B, C y D se han asociado con la formación de tumores.
La actividad del complejo succinato deshidrogenasa puede ser regulada por modificaciones postraduccionales como la fosforilación y la acetilación, aunque también puede ocurrir inhibición del sitio activo.
La acetilación de algunos residuos de lisina puede disminuir la actividad de esta enzima y este proceso es llevado a cabo por una enzima deacetilasa conocida como SIRT3; la fosforilación tiene el mismo efecto sobre la enzima.
Además de estas modificaciones, el complejo SDH también es regulado por los intermediarios del ciclo de Krebs, concretamente el oxalacetato y el succinato. El oxalacetato es un potente inhibidor, mientras que el succinato favorece la disociación del oxalacetato, funcionando como activador.
La deficiencia de succinato deshidrogenasa es una anomalía o trastorno de la cadena respiratoria mitocondrial. Esta deficiencia es causada por las mutaciones de los genes SDHA (o SDHAF1), SDHB, SDHC y SDHD.
Diferentes investigaciones han demostrado mutaciones homocigotas y heterocigotas en estos genes, en especial del SDHA. Las mutaciones de estos genes provocan sustituciones de aminoácidos en la proteína (en cualquiera de las subunidades SDHA, B, C o D), o en su defecto codifican proteínas anormalmente cortas.
Por consiguiente, las sustituciones de aminoácidos y las codificaciones de proteínas anormalmente cortas, dan lugar a trastornos o alteraciones de la enzima SDH, que provocan una falla en la capacidad óptima de las mitocondrias para producir energía. Esto es lo que los científicos llaman como trastorno de la cadena respiratoria mitocondrial.
Este trastorno se puede expresar en el ser humano fenotípicamente de muchas formas. Las más conocidas son: deficiencia o falta del desarrollo lingüístico, cuadriplejia espástica, contracciones involuntarias musculares (distonía), debilidad muscular, y miocardiopatias, entre otros problemas relacionados.
Algunos pacientes con deficiencia de succinato deshidrogenasa pueden llegar a manifestar la enfermedad de Leigh o el síndrome de Kearns-saire.
Ciertos estudios sugieren el uso de pruebas y análisis histoquímicos cualitativos, así como análisis bioquímicos cuantitativos, enzimáticos, de la cadena respiratoria. Otros por su parte sugieren la ampliación completa mediante la reacción en cadena de la polimerasa (PCR) de los exones de las subunidades en estudio y luego, la respectiva secuenciación.
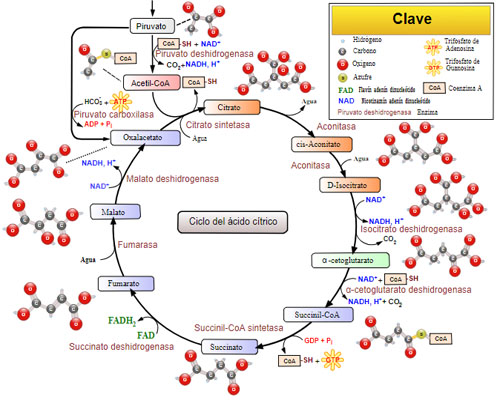
Existe una gran cantidad de expresiones fenotípicas producidas por trastornos de la cadena respiratoria mitocondrial, debido a la deficiencia de succinato deshidrogenasa. Sin embargo, cuando de síndromes o enfermedades se trata, se habla de las siguientes.
Es una enfermedad neurológica progresiva, asociada a mutaciones en el genoma nuclear (en este caso de la succinato deshidrogenasa), que afectan el complejo piruvato-deshidrogenasa hasta la ruta de la fosforilación oxidativa.
Los síntomas aparecen antes del primer año de edad del individuo, pero en casos atípicos, se han observado los primeros síntomas durante la adolescencia.
Entre los síntomas más comúnmente observados son: hipotonía con pérdida de control cefálico, movimientos involuntarios, vómitos recurrentes, problemas respiratorios, incapacidad para mover el glóbulo ocular, signos piramidales y extrapiramidales entre otros. Las convulsiones no son muy comunes.
Es posible que la enfermedad pueda ser detectada en diagnósticos prenatales. No se conoce una cura o un tratamiento especifico, pero algunos especialistas sugieren tratamientos con ciertos vitamínicos o cofactores.
Comúnmente llamado GIST, es un tipo de tumor del tracto gastrointestinal, que generalmente se desarrolla en áreas como el estómago o el intestino delgado. Se cree que la causa de estos se debe a un cierto grupo de células altamente especializadas denominadas células ICC o células intersticiales de Cajal.
Otras consideraciones sobre la causa de los GIST, son las mutaciones de ciertos tipos de genes, que según algunos autores causan el 90% de los tumores. Los genes implicados son: genes KIT, PDGFRA, succinato deshidrogenasa (SDH) – deficiente.
La succinato deshidrogenasa (SDH) – deficiente, ocurre principalmente en mujeres jóvenes, produce tumores en el estómago y con relativa frecuencia produce metástasis en los ganglios linfáticos. Un pequeño porcentaje ocurre en niños y en la mayoría de los casos, se debe a la falta de expresión de la subunidad SDHB.
Se ha determinado que algunos pacientes con deficiencias de succinato deshidrogenasa, pueden manifestar el síndrome Kearns-Sayre. Esta enfermedad está relacionada con trastornos mitocondriales, y se caracteriza por la ausencia de movimiento de los globos oculares.
Otras características de esta enfermedad son la retinitis pigmentaria, sordera, cardiomiopatía y afecciones del sistema nerviosos central. Usualmente estos síntomas se observan antes de que el paciente cumpla los 20 años de edad. No se conoce ningún diagnóstico prenatal para esta afección.
Tampoco se conoce cura para esta enfermedad. El tratamiento es paliativo, es decir, solo funciona para reducir los efectos de la enfermedad, no la cura. Por otra parte, aunque depende del número de órganos afectados y de la atención medica recibida, la esperanza de vida es relativamente normal.
- Ackrell, B. A., Kearney, E. B., & Singer, T. P. (1978). [47] Mammalian succinate dehydrogenase. In Methods in enzymology (Vol. 53, pp. 466-483). Academic Press.
- Brière, J. J., Favier, J., Ghouzzi, V. E., Djouadi, F., Benit, P., Gimenez, A. P., & Rustin, P. (2005). Succinate dehydrogenase deficiency in human. Cellular and Molecular Life Sciences CMLS, 62(19-20), 2317-2324.
- Cecchini, G., Schröder, I., Gunsalus, R. P., & Maklashina, E. (2002). Succinate dehydrogenase and fumarate reductase from Escherichia coli. Biochimica et Biophysica Acta (BBA)-Bioenergetics, 1553(1-2), 140-157.
- Hatefi, Y., & Davis, K. A. (1971). Succinate dehydrogenase. I. Purification, molecular properties, and substructure. Biochemistry, 10(13), 2509-2516.
- Hederstedt, L. A. R. S., & Rutberg, L. A. R. S. (1981). Succinate dehydrogenase–a comparative review. Microbiological reviews, 45(4), 542.
- Nelson, D. L., Lehninger, A. L., & Cox, M. M. (2008). Lehninger principles of biochemistry. Macmillan.
- Rutter, J., Winge, D. R., & Schiffman, J. D. (2010). Succinate dehydrogenase–assembly, regulation and role in human disease. Mitochondrion, 10(4), 393-401.