Glucógeno: estructura, síntesis, degradación, funciones
El glucógeno es el carbohidrato de almacenamiento de la mayor parte de los mamíferos. Los carbohidratos son comúnmente llamados azúcares y estos se clasifican de acuerdo al número de residuos originados por hidrólisis (monosacáridos, disacáridos, oligosacáridos y polisacáridos).
Los monosacáridos son los carbohidratos más simples que se clasifican de acuerdo al número de carbonos contenidos en su estructura. Existen entonces las triosas (3C), tetrosas (4C), pentosas (5C), hexosas (6C), heptosas (7C) y octosas (8C).
Según la presencia del grupo aldehído o del grupo cetona, estos monosacáridos se clasifican también en aldosas o cetosas respectivamente.
Los disacáridos dan origen, por hidrólisis, a dos monosacáridos simples, mientras que los oligosacáridos producen de 2 a 10 unidades de monosacáridos y los polisacáridos producen más de 10 monosacáridos.
El glucógeno es, desde el punto de vista bioquímico, un polisacárido compuesto por cadenas ramificadas de una aldosa de seis carbonos, es decir, una hexosa conocida como glucosa. Gráficamente puede representarse al glucógeno como un árbol de glucosa. Este también es llamado el almidón animal.
La glucosa en las plantas se almacena como almidón y en los animales como glucógeno, que se almacena primordialmente en el hígado y en el tejido muscular.
En el hígado, el glucógeno puede llegar a constituir el 10% de su masa y el 1% de la masa muscular. Como en un hombre de 70Kg el hígado pesa unos 1800 g y los músculos unos 35 Kg, la cantidad total de glucógeno muscular es mucho mayor que la hepática.
Índice del artículo
- 1 Estructura
- 2 Síntesis
- 3 Degradación
- 4 Regulación de la síntesis y la degradación
- 5 Funciones
- 6 Enfermedades relacionadas
- 7 Referencias
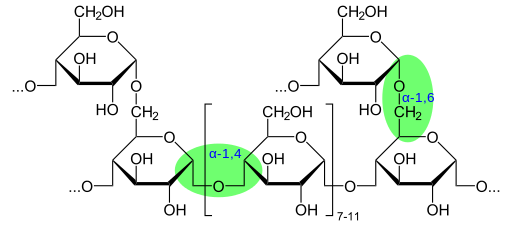
El peso molecular del glucógeno puede llegar a 108 g/mol, que equivalen a 6×105 moléculas de glucosa. El glucógeno está formado por múltiples cadenas ramificadas de α-D-glucosa. La glucosa (C6H12O6) es una aldohexosa que puede representarse en forma lineal o cíclica.
El glucógeno posee una estructura muy ramificada y compacta con cadenas de 12 a 14 residuos de glucosa en forma de α-D-glucosa que se enlazan con enlaces α-(1→4) glucosídicos. Las ramificaciones de la cadena se forman mediante enlaces α-(1→6) glucosídicos.
El glucógeno, al igual que el almidón que se ingiere en la dieta, aporta la mayor parte de los carbohidratos que el cuerpo necesita. En el intestino estos polisacáridos son degradados por hidrólisis y luego absorbidos hacia el torrente circulatorio principalmente como glucosa.
Tres enzimas: la ß-amilasa, la α-amilasa y la amilo-α-(1→6)-glucosidasa son responsables de la degradación intestinal tanto del glucógeno como del almidón.
La α-amilasa hidroliza al azar los enlaces α-(1→4) de las cadenas laterales tanto del glucógeno como del almidón, y por ello recibe el nombre de endoglicosidasa. La ß-amilasa es una exoglicosidasa que va liberando dímeros de ß-maltosa rompiendo enlaces α-(1→4) glicosídicos desde los extremos de las cadenas más externas sin llegar a las ramificaciones.
En vista de que ni la ß-amilasa ni la α-amilasa degradan los puntos de ramificación, el producto final de su acción es una estructura altamente ramificada de unos 35 a 40 residuos de glucosa que se denomina dextrina límite.
La dextrina límite finalmente es hidrolizada en los puntos de ramificación que poseen enlaces α-(1→6) por medio de la amilo-α-(1→6)-glucosidasa, conocida también como enzima “desramificadora”. Las cadenas liberadas por esta desramificación son luego degradadas por la ß-amilasa y la α-amilasa.
Como el glucógeno ingerido ingresa como glucosa, el que se encuentra en los tejidos deberá ser sintetizado por el organismo a partir de glucosa.
La síntesis de glucógeno recibe el nombre de glucogénesis y tiene lugar sobre todo en el músculo y el hígado. La glucosa que ingresa al organismo con la dieta pasa al torrente circulatorio y de allí al interior de las células, donde inmediatamente es fosforilada por acción de una enzima llamada glucoquinasa.
La glucoquinasa fosforila a la glucosa en el carbono 6. El ATP proporciona el fósforo y la energía para esta reacción. Como resultado, se forma la glucosa 6-fosfato y se libera un ADP. Luego, la glucosa 6-fosfato se convierte en glucosa 1-fosfato por la acción de una fosfoglucomutasa que muda el fósforo de la posición 6 a la posición 1.
La glucosa 1-fosfato queda activada para la síntesis de glucógeno, que implica la participación de un conjunto de otras tres enzimas: la UDP-glucosa pirofosforilasa, la glucógeno sintetasa y la amilo-(1,4→1,6)-glicosiltransferasa.
La glucosa-1-fosfato, junto con la uridina trifosfato (UTP, un nucleósido de trifosfato de uridina) y por acción de la UDP-Glucosa-pirofosforilasa, forma el complejo uridina difosfato-glucosa (UDP Glc). En el proceso se hidroliza un ion pirofosfato.
Después, la enzima glucógeno sintetasa forma un enlace glucosídico entre el C1 del complejo UDP Glc y el C4 de un residuo terminal de glucosa del glucógeno, y se libera el UDP del complejo activado de glucosa. Para que esta reacción ocurra debe existir una molécula preexistente de glucógeno llamada “glucógeno primordial”.
El glucógeno primordial se sintetiza sobre una proteína cebadora, la glucogenina, que tiene 37 kDa y que se glicosila en un residuo de tirosina mediante el complejo UDP Glc. A partir de allí se van enlazando residuos de α-D-Glucosa con enlaces 1→4 y se forma una pequeña cadena sobre la cual actúa la glucógeno sintetasa.
Una vez que la cadena inicial enlaza como mínimo 11 residuos de glucosa, la enzima ramificante o amilo-(1,4→1,6)-glicosiltransferasa transfiere un pedazo de cadena de 6 o 7 residuos de glucosa a la cadena adyacente en posición 1→6, con lo cual se establece un punto de ramificación. La molécula de glucógeno así construida va creciendo por adiciones de unidades de glucosa con enlaces glucosídicos 1→4 y más ramificaciones.
La degradación del glucógeno recibe el nombre de glucogenólisis, y no es equivalente a la vía reversa de su síntesis. La velocidad de esta ruta está limitada por la velocidad de la reacción catalizada por la glucógeno fosforilasa.
La glucógeno fosforilasa se encarga de la escisión (fosforólisis) de los enlaces 1→4 de las cadenas de glucógeno, liberando glucosa 1-fosfato. La acción enzimática se inicia en los extremos de las cadenas más externas y se remueven secuencialmente hasta que quedan 4 residuos de glucosa a cada lado de las ramificaciones.
Luego, otra enzima, la α-(1→4) → α-(1→4) glucano transferasa, deja expuesto el punto de ramificación al transferir una unidad de trisacárido de una rama a otra. Esto permite que la amilo-(1→6)-glucosidasa (enzima desramificante) hidrolice el enlace 1→6 quedando removida la rama que sufrirá la acción de la fosforilasa. La acción combinada de estas enzimas termina escindiendo completamente al glucógeno.
Como la reacción inicial de la fosfomutasa es reversible, se puede formar glucosa 6-fosfato a partir de los residuos de glucosa 1-fosfato escindidos del glucógeno. En el hígado y en el riñón, pero no en el músculo, existe una enzima, la glucosa-6-fosfatasa, capaz de desfosforilar a la glucosa 6-fosfato y convertirla en glucosa libre.
La glucosa defosforilada puede difundir a la sangre, y es así como la glucogenólisis hepática se refleja en un aumento de los valores de glucosa sanguínea (glicemia).
Este proceso se ejerce sobre dos enzimas fundamentales: la glucógeno sintetasa y la glucógeno fosforilasa, de manera tal que cuando una de ellas está activa la otra está en su estado inactivo. Esta regulación evita que reacciones opuestas de síntesis y degradación ocurran simultáneamente.
La forma activa y la forma inactiva de ambas enzimas es muy diferente, y la interconversión de las formas activas e inactivas de la fosforilasa y de la glucógeno sintetasa está sometida a un estricto control hormonal.
La adrenalina es una hormona que se libera desde la médula suprarrenal, y el glucagón es otra que se produce en la parte endocrina del páncreas. El páncreas endocrino produce insulina y glucagón. Las células α de los islotes de Langerhans son las que sintetizan el glucagón.
Adrenalina y glucagón son dos hormonas que se liberan cuando se necesita energía en respuesta a la disminución de los niveles de glucosa en sangre. Estas hormonas estimulan la activación de la glucógeno fosforilasa e inhiben a la glucógeno sintetasa, estimulando así la glucogenólisis e inhibiendo la glucogénesis.
Mientras que la adrenalina ejerce su acción sobre el músculo y el hígado, el glucagón sólo actúa sobre el hígado. Estas hormonas se unen a receptores membranales específicos en la célula blanco, lo que activa a la adenilato ciclasa.
La activación de la adenilato ciclasa inicia una cascada enzimática que, por un lado, activa a una proteinquinasa dependiente de AMPc que inactiva a la glucógeno sintetasa y activa a la glucógeno fosforilasa por fosforilación (directa e indirectamente, respectivamente).
El músculo esquelético posee otro mecanismo de activación de la glucógeno fosforilasa a través del calcio, que se libera como consecuencia de la despolarización de la membrana muscular al inicio de la contracción.
Las cascadas enzimáticas anteriormente descritas terminan aumentando los niveles de glucosa y cuando estos alcanzan cierto nivel, se activa la glucogénesis y se inhibe la glucogenólisis, inhibiéndose también la ulterior liberación de adrenalina y glucagón.
La glucogénesis se activa por medio de la activación de la fosforilasa fosfatasa, una enzima que regula la síntesis de glucógeno por varios mecanismos, que implican la inactivación de la fosforilasa quinasa y de la fosforilasa α, que es un inhibidor de la glucógeno sintetasa.
La insulina promueve el ingreso de la glucosa al interior de las células musculares, aumentando los niveles de glucosa 6-fosfato, que estimula la desfosforilación y activación de la glucógeno sintetasa. Así se inicia la síntesis y se inhibe la degradación del glucógeno.
El glucógeno muscular constituye una reserva energética para el músculo que, al igual que las grasas de reserva, le permiten al músculo cumplir con sus funciones. Siendo una fuente de glucosa, el glucógeno muscular se utiliza durante el ejercicio. Estas reservas aumentan con el entrenamiento físico.
En el hígado, el glucógeno también constituye una fuente importante de reserva tanto para las funciones del órgano como para el aporte de glucosa al resto del cuerpo.
Esta función del glucógeno hepático se debe al hecho de que el hígado contiene glucosa 6-fosfatasa, una enzima capaz de eliminar el grupo fosfato de la glucosa 6-fosfato y convertirla en glucosa libre. La glucosa libre, a diferencia de la glucosa fosforilada, puede difundirse a través de la membrana de los hepatocitos (las células del hígado).
Es así como el hígado puede aportar glucosa a la circulación y mantener los niveles de glucosa estables, aún en condiciones de ayuno prolongado.
Esta función es de gran importancia, ya que el cerebro se nutre casi exclusivamente de la glucosa sanguínea, por lo que las hipoglucemias severas (muy bajas concentraciones de glucosa en sangre) pueden producir pérdida de conocimiento.
Las enfermedades relacionadas con el glucógeno reciben el nombre genérico de “enfermedades por almacenamiento de glucógeno”.
Estas enfermedades constituyen un grupo de patologías hereditarias caracterizadas por el depósito en los tejidos de cantidades o tipos anormales de glucógeno.
La mayor parte de las enfermedades de almacenamiento de glucógeno se producen por un déficit de naturaleza genética de cualquiera de las enzimas involucradas en el metabolismo del glucógeno.
Se clasifican en ocho tipos, la mayoría de los cuales tienen nombres propios y cada una de ellas se produce por un déficit enzimático distinto. Algunas son mortales en etapas muy tempranas de la vida, mientras que otras se acompañan de debilidad muscular y déficit durante el ejercicio.
Algunas de las enfermedades relacionadas con el glucógeno más destacadas son las siguientes:
– La enfermedad de Von Gierke o enfermedad por almacenamiento de glucógeno de Tipo I, se produce por un déficit de glucosa 6-fosfatasa en hígado y el riñón.
Está caracterizada por el crecimiento anormal del hígado (hepatomegalia) debido a la acumulación exagerada de glucógeno e hipoglucemia, ya que el hígado se hace incapaz de aportar glucosa a la circulación. Los pacientes con esta condición presentan alteraciones de crecimiento.
– La enfermedad de Pompe o de Tipo II, se debe a un déficit de la α-(1→4)-glucano 6-glicosiltransferas en el hígado, el corazón y los músculos esqueléticos. Esta enfermedad, al igual que la de Andersen o de Tipo IV, es letal antes de los dos años de vida.
– La enfermedad de McArdle o de Tipo V, presenta un déficit de fosforilasa muscular y se acompaña de debilidad muscular, disminución de la tolerancia al ejercicio, acumulación anormal de glucógeno muscular y ausencia de lactato durante el ejercicio.
- Bhattacharya, K. (2015). Investigation and management of the hepatic glycogen storage diseases. Translational Pediatrics, 4(3), 240–248.
- Dagli, A., Sentner, C., & Weinstein, D. (2016). Glycogen Storage Disease Type III. Gene Reviews, 1–16.
- Guyton, A., & Hall, J. (2006). Textbook of Medical Physiology (11th ed.). Elsevier Inc.
- Mathews, C., van Holde, K., & Ahern, K. (2000). Biochemistry (3th ed.). San Francisco, California: Pearson.
- Mckiernan, P. (2017). Pathobiology of the Hepatic Glycogen Storage Diseases. Curr Pathobiol Rep.
- Murray, R., Bender, D., Botham, K., Kennelly, P., Rodwell, V., & Weil, P. (2009). Harper’s Illustrated Biochemistry (28th ed.). McGraw-Hill Medical.
- Nelson, D. L., & Cox, M. M. (2009). Lehninger Principios de Bioquímica. Ediciones Omega (5th ed.).
- Rawn, J. D. (1998). Biochemistry. Burlington, Massachusetts: Neil Patterson Publishers.
- Tarnopolsky, M. A. (2018). Myopathies Related to Glycogen Metabolism Disorders. Neurotherapeutics.