Síndrome de Potter: qué es, causas, síntomas, tratamiento
¿Qué es el síndrome de Potter?
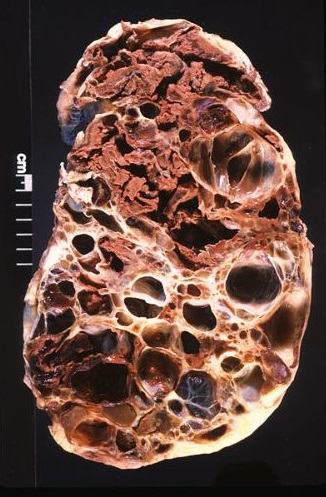
El síndrome de Potter es un raro y grave trastorno de tipo hereditario autosómico recesivo que afecta a recién nacidos y se caracteriza por oligohidramnios acentuado (falta de líquido amniótico), riñones poliquísticos, agenesia renal y uropatía obstructiva.
Esta enfermedad fue descrita por primera vez por el patólogo Edith Potter en 1946, que señalaba las características faciales similares de una serie de bebés que tenían agenesia renal bilateral. A partir de ahí, fue desentrañando poco a poco los síntomas típicos de la enfermedad.
También se ha llamado secuencia de Potter o secuencia de oligohidramnios. El concepto del síndrome de Potter al principio solo se refería a los casos provocados por agenesia renal bilateral, aunque actualmente muchos investigadores lo usan para cualquier caso que aparezca unido a falta de líquido amniótico.
Causas
La producción de orina en el feto es el mecanismo principal para producir un volumen adecuado de líquido amniótico, que comienza sobre el cuarto mes de embarazo.
El feto continuamente traga líquido amniótico, este se vuelve a absorber en el intestino y se expulsa de nuevo a través de los riñones (por la orina) a la cavidad amniótica.
En esta enfermedad, la cantidad de líquido amniótico es insuficiente principalmente porque no funcionan bien los riñones del bebé. Normalmente, lo que ocurre es que en el periodo de gestación no se forman los riñones adecuadamente, faltando uno o ambos (agenesia renal).
Aunque también puede suceder la obstrucción de las vías urinarias o, algunas veces, la rotura de la membrana que encierra el líquido amniótico. Esta falta de líquido amniótico es el principal causante de los síntomas del síndrome de Potter.
Genética
La enfermedad de Potter puede ocurrir por dos enfermedades genéticas, que son tanto la enfermedad renal poliquística autosómica dominante como la autosómica recesiva. De esta manera, los antecedentes familiares de enfermedades de los riñones pueden aumentar el peligro de desarrollar este síndrome en el feto.
Así, en casos de familias con historia de agenesia renal unilateral o bilateral, esto puede suponer un rasgo autosómico dominante.
Aunque se han asociado ciertas mutaciones genéticas a condiciones presentadas comúnmente en el síndrome de Potter, como la enfermedad autosómica recesiva o dominante de los riñones poliquísticos y la displasia renal multiquística, no se encuentra nada definitivo en la agenesia renal bilateral.
En resumen, los rasgos genéticos concretos no se conocen con certeza hoy en día y es algo que se continúa investigando.
Lo que sí se sabe es que parece no existir una asociación directa del abuso de sustancias o factores ambientales peligrosos durante el embarazo en la aparición de la agenesia renal bilateral o en el síndrome de Potter.
Síntomas
Los principales síntomas son:
– Insuficiencia renal.
– Carencia de líquido amniótico: lo que puede provocar muchos problemas, ya que el líquido ayuda a lubricar las partes del cuerpo del feto, lo protege y contribuye al desarrollo de sus pulmones. Cuando falta este líquido, la cavidad amniótica es más pequeña de lo normal y acaba dejando poco espacio al feto, lo que impide su crecimiento normal.
– Nacimiento prematuro.
– Malformaciones: sobre todo en las extremidades inferiores, como en los pies y arqueamiento de las piernas. También puede llegar a presentarse sirenomelia, o síndrome de sirena, que consiste en la fusión de las piernas.
– Apariencia facial atípica como puente nasal ancho o nariz “en pico de loro”, ojos separados y orejas situadas más abajo de lo normal.
– Exceso de piel, siendo frecuente en los afectados un pliegue cutáneo en la zona de la mejilla.
– Glándulas suprarrenales con apariencia de pequeños discos ovalados que presionan el abdomen posterior asociado al mal funcionamiento renal.
– Vejiga más pequeña de lo normal y poco dilatable, almacenando muy poco fluido.
– En los hombres pueden faltar el conducto deferente y las vesículas seminales.
– En las mujeres puede que no se desarrolle el útero y la parte superior de la vagina.
– Atresia anal: se produce cuando no se conecta correctamente el recto con el ano. Puede ocurrir lo mismo en el esófago, duodeno o la arteria umbilical.
– A veces se puede dar una hernia diafragmática congénita que impide el buen desarrollo del diafragma.
– Pulmones inmaduros o hipoplasia pulmonar (anomalía congénita caracterizada por una interrupción del desarrollo pulmonar). Este mecanismo no está del todo claro, aunque parece ser que influye el adecuado movimiento del líquido amniótico a través de los pulmones durante la etapa fetal. Evidentemente, si no hay suficiente líquido amniótico, los pulmones no se van a desarrollar correctamente.
– Consecuencia de lo anterior, son graves problemas respiratorios, que suelen ser los causantes de la mortalidad temprana en los afectados.
Trastornos asociados
Además de los ya mencionados, el síndrome de Potter se ha relacionado con otros problemas tales como síndrome de Down, el síndrome de Kallmann, y el síndrome branquio-oto-renal (BOR), entre otros.
¿Cómo se diagnostica?
Durante el embarazo puede verse a través de ecografías si existe menos líquido amniótico de la cuenta, o si el feto presenta anormalidades en los riñones o ausencia de estos.
Para detectar los posibles problemas en el recién nacido, puede ser necesario hacer una radiografía de los pulmones y del abdomen.
Por otro lado, se puede acudir a un asesor genético que va a tomar una muestra de sangre en el feto para llevar a cabo una amniocentesis. Esto sirve para mirar si el número de cromosomas es correcto o si existen alteraciones en algunas de sus partes o translocaciones.
Puede ser útil para descartar otras enfermedades asociadas como el síndrome de Down. Para detectar las posibles mutaciones heredadas es esencial la exploración del genoma del padre, madre, bebé afectado y hermanos.
Tratamiento
No existen tratamientos para esta enfermedad y su pronóstico es muy negativo, suelen morir antes de nacer o al poco tiempo después. Si sobrevive en el nacimiento, puede que sea necesaria la reanimación.
También se pueden usar algunos métodos para paliar los síntomas y mejorar dentro de lo posible la vida, como trasplante de órganos o intervención para la uropatía obstructiva.
Sin embargo, existe un caso de una bebé con síndrome de Potter nacida en julio de 2013, cuya madre es una congresista norteamericana, Jaime Herrera Beutler, que vive en la actualidad. Esto es porque unas semanas antes de nacer se inyectó una solución salina en el útero materno con el objetivo de ayudar al desarrollo pulmonar del feto.
Al nacer el bebé se comprobó que la intervención había sido un éxito y que podía respirar por sí misma. Su padre le donó un riñón, y a día de hoy, Abigail es una niña de 9 años que a pesar de todo está creciendo, dentro de su condición, de una forma adecuada.
Referencias
- Oligohydramnios Sequence (Potter’s Syndrome). Obtenido de Healthline.
- Potter Syndrome. Obtenido de Medscape.