Síndrome de Apert: qué es, síntomas, causas y tratamiento
¿Qué es el síndrome de Apert?
El síndrome de Apert, o acrocefalosindactilia tipo I (ACS1), es una patología de origen genético que se caracteriza por la presencia de diferentes alteraciones y malformaciones en el cráneo, la cara y las extremidades.
A nivel clínico, el síndrome de Apert se caracteriza por la presencia o desarrollo de un cráneo puntiagudo o alargado, área facial hundida con una alteración en la proyección de las piezas dentales, fusión y cierre de los huesos de dedos y articulaciones, retraso mental variable, alteraciones de lenguaje, etc.
A pesar de que esta patología puede ser hereditaria, en la mayoría de los casos el síndrome de Apert ocurre sin la presencia de antecedentes familiares, debido esencialmente a una mutación de novo durante la fase de gestación.
Los mecanismos genéticos que provocan el síndrome de Apert no se conocen con exactitud. Actualmente, se han identificado diversas alteraciones genéticas capaces de producir esta patología, relacionadas esencialmente con mutaciones en el gen FGFR2.
Signos y síntomas
Las manifestaciones clínicas del síndrome de Apert suelen incluir una malformación o desarrollo incompleto de la estructura craneal, un fenotipo o patrón facial atípico y alteraciones esqueléticas en las extremidades.
En el caso del síndrome de Apert, la afectación central está relacionada con la formación y cierre de la estructura ósea del cráneo. Durante el desarrollo embrionario, tiene lugar un proceso denominado craneosinostosis, caracterizado por un cierre prematuro de las suturas craneales.
Las fisuras o suturas craneales son un tipo de bandas de tejido fibroso que tienen el objetivo fundamental de conectar los huesos que conforman el cráneo (frontal, occipital, parietal y temporal).
Durante la fase de gestación y el periodo posnatal temprano, la estructura ósea que conforma el cráneo se mantiene unida gracias a estos tejidos fibrosos y elásticos.
Normalmente, los huesos craneales no suelen fusionarse hasta los 12 o 18 meses aproximadamente. La presencia de espacios o puntos blandos entre los huesos craneales es parte del desarrollo infantil normal.
Por lo tanto, durante toda la etapa infantil, estas suturas o regiones flexibles permiten al cerebro crecer de forma acelerada y, además, lo protege de impactos.
En el síndrome de Apert, el cierre prematuro de estas suturas craneales y de los huesos craneales imposibilita el desarrollo normal del crecimiento craneal y cerebral.
Los signos y síntomas más frecuentes del síndrome de Apert pueden incluir:
Alteraciones y anomalías craneofaciales
– Craneosinostosis: el cierre temprano de las suturas del cráneo provoca una amplia variedad de alteraciones craneofaciales, entre las que pueden incluirse la expansión inadecuada de las estructuras cerebrales, desarrollo de edema papilar (inflamación del punto ciego ocular donde surge el nervio óptico), atrofia óptica (lesión o déficit que afecta a la funcionalidad ocular) y/o hipertensión intracraneal (incremento anormal de la presión del líquido cefalorraquídeo).
– Hipoplasia facial unilateral o bilateral: la cabeza presenta una apariencia atípica con un desarrollo deficiente o incompleto de alguna de sus mitades. A nivel visual, se observa un rostro hundido, con ojos sobresalientes y párpados caídos.
– Proptosis o exoftalmos: protrusión significativa y anormal de los ojos hacia el exterior de la cavidad ocular.
– Macroglosia: incremento del tamaño de la lengua debido a la presencia de un volumen de tejido superior a lo normal.
– Maloclusión mandibular: son frecuentes la presencia de diferentes alteraciones relacionadas con el crecimiento de la estructura ósea de la mandíbula, que impiden el correcto funcionamiento y cierre del sistema o aparato masticatorio.
– Fisura palatina: presencia de un agujero/fisura en el área central o media del paladar.
Alteraciones y anomalías músculo-esqueléticas
Este tipo de alteraciones afectan fundamentalmente a las extremidades superiores e inferiores, normalmente a la fusión y desarrollo de los dedos.
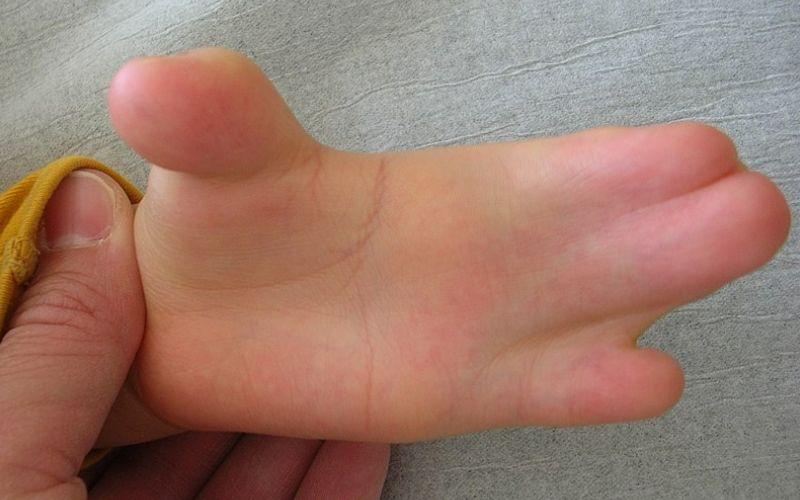
– Sindactilia: fusión anormal y patológica de uno o varios dedos entre sí, en las manos o en los pies. Pueden distinguirse diversas variantes, tipo I (fusión del 2º, 2º y 4º dedo), tipo II (fusión 5º dedo), tipo III (fusión de todos los dedos). Generalmente, en las manos son más frecuentes las sindactilias de tipo I, mientras que en los pies, son más frecuentes las de tipo III.
Además de estas, también es posible observar otros hallazgos clínicos a nivel músculo-esquelético: acortamiento de diversos huesos (radio, húmero, fémur), hipoplasia de la escápula o la pelvis, fusión de vértebras cervicales.
Como consecuencia, muchos afectados van a presentar una movilidad articular reducida y, por tanto, podrán desarrollar diversas dificultadas para la adquisición de la motricidad gruesa y fina.
Alteraciones y anomalías cutáneas/dermatológicas
Este tipo de anomalías son muy heterogéneas y variables entre los individuos afectados, sin embargo, se han identificado algunas de las más comunes:
– Hiperhidrosis: incremento excesivo de la sudoración, especialmente en manos y pies.
– Lesiones maculo-vesiculosas o costrosas: lo más frecuente es la presencia de lesiones cutáneas de tipo acneiforme.
– Hipopigmentación: cambios en el color de la piel que implican una disminución de la pigmentación.
– Engrosamiento cutáneo: incremento anormal del grosor de la piel en una o varias áreas.
Alteraciones y anomalías viscerales
La alteración etiológica de esta patología puede dar lugar al desarrollo de lesiones o patologías secundarias a nivel morfológico y estructural en diversas áreas corporales, algunas de ellas incluyen:
– Malformación en el sistema nervioso central: se ha observado en algunos casos el desarrollo de agenesia o hipoplasia del cuerpo calloso (ausencia o desarrollo parcial) y de diversas estructuras del sistema líbico. Además, también se ha descrito un desarrollo anormal o alterado de la sustancia blanca cerebral.
– Malformaciones genitourinarias: en el caso de los afectados varones, pueden aparecer válvulas uretrales posteriores que ocasionen insuficiencia renal e hidronefrosis. Por otro lado, en el caso de las mujeres afectadas, es frecuente la presencia de malformaciones en el clítoris.
– Malformaciones cardiacas: las alteraciones relacionadas con la función cardiaca y el corazón suelen estar asociadas a la presencia de hipoplasia ventricular izquierda o comunicación intraventricular.
Alteraciones y anomalías cognitivas/psicológicas
A pesar de que, en muchos casos, es posible observar la presencia de una alteración general de las funciones cognitivas y del nivel intelectual, el retraso mental no está presente de forma inequívoca en todos los casos de síndrome de Apert.
Además, en los casos en los que sí existe una afectación del nivel intelectual, esta puede ser variable, en una escala de leve a moderada.
En el área lingüística, es frecuente el desarrollo de diversos déficits, relacionados fundamentalmente con la articulación de los sonidos producto de las malformaciones mandibulares y bucales.
Causas
El síndrome de Apert se debe a la presencia de una mutación específica en el gen FGFR2. Los estudios experimentales han señalado que este gen se encarga de la producción de una proteína, denominada receptor 2 del factor de crecimiento de fibroblastos.
Entre las funciones de este factor, se describe el envío de diferentes señales químicas hacia células inmaduras para ocasionar su transformación y diferenciación en células óseas durante la fase de desarrollo fetal o prenatal.
Por lo tanto, la presencia de mutaciones en el gen FGFR2 altera el funcionamiento de esta proteína y, en consecuencia, puede causar la fusión temprana de los huesos del cráneo, manos y pies.
Diagnóstico
Buena parte de los rasgos clínicos del síndrome de Apert pueden identificarse durante la gestación, específicamente en las ecografías de control del embarazo y de desarrollo fetal.
De esta forma, cuando existe una sospecha clínica, se recomienza la realización de un estudio genético para identificar la presencia de una mutación genética compatible con el síndrome de Apert.
Por otro lado, cuando los signos son sutiles o no se han identificado antes del nacimiento, tras este es posible realizar un análisis físico pormenorizado y diversas pruebas genéticas para confirmar el diagnóstico.
¿Existe tratamiento para el síndrome de Apert?
A pesar de que no existe una cura específica para el síndrome de Apert, se han descrito diversos abordajes para el tratamiento de los síntomas y complicaciones médicas propias de esta patología.
Las intervenciones terapéuticas más eficaces son aquellas que se implementan de forma temprana, en los primeros momentos de vida e implican a profesionales de diferentes áreas.
Normalmente, el tratamiento de los niños afectados requiere una planificación individualizada, con la programación de múltiples cirugías. Así, el manejo de esta patología se basa en la corrección de las malformaciones esqueléticas y cráneo-faciales, y el apoyo psicológico y neuropsicológico.
A través de la neurocirugía se persigue reconstruir la bóveda craneal, mientras que los especialistas en la cirugía maxilofacial tratan de corregir las malformaciones faciales. Por otro lado, también es frecuente la participación de cirujanos de trauma, para la reconstrucción de las malformaciones presentes en manos y pies.
Además, el diseño de programas individualizados de estimulación temprana, rehabilitación de la comunicación, entrenamiento de habilidades sociales o el seguimiento psicopedagógico, resultan beneficiosos para la consecución de un desarrollo óptimo, funcional e independiente de los individuos afectados.
Referencias
- Apert syndrome. Obtenido de Boston Childre’s Hospital.
- Apert syndrome. Obtenido de Genetics Home Reference.
- Apert Syndrome. Obtenido de MedlinePlus.