Árbol filogenético: tipos y sus características, ejemplos
Un árbol filogenético es una representación gráfica matemática de la historia y de las relaciones ancestro – descendientes de grupos, poblaciones, especies, o cualquier otra categoría taxonómica. Teóricamente, todos los árboles filogenéticos pueden agruparse en el árbol de la vida, constituyendo el árbol universal.
Estas representaciones gráficas han revolucionado el estudio de la biología evolutiva, ya que permiten establecer y definir una especie, probar diversas hipótesis evolutivas (como la teoría endosimbiótica), evaluar el origen de enfermedades (como el VIH), etc.
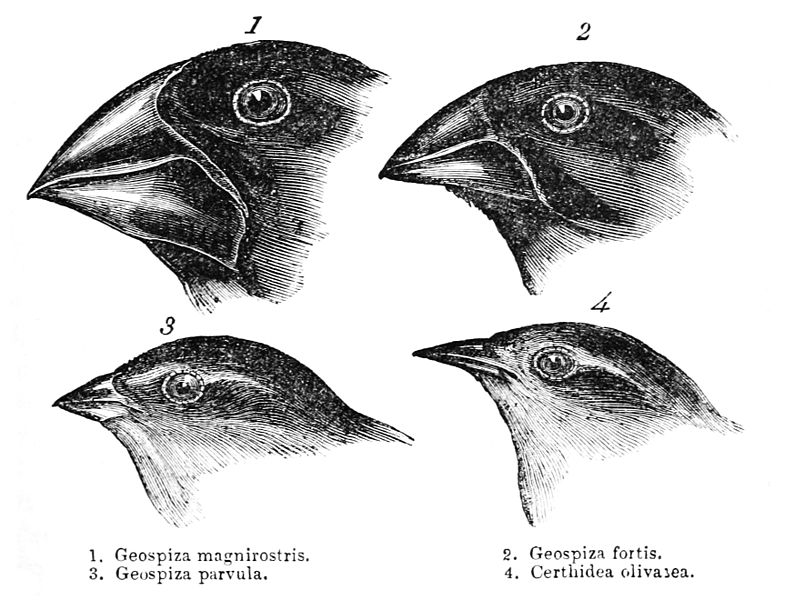
Los árboles pueden reconstruirse usando caracteres morfológicos o moleculares, o ambos. Del mismo modo, existen diversos métodos para construirlos, siendo el más común la metodología cladista. Esta busca identificar los caracteres derivados compartidos, conocidos como sinapomorfia.
Índice del artículo
- 1 Características
- 2 Tipos
- 3 Errores más comunes al leer los árboles filogenéticos
- 4 Ejemplos
- 5 Referencias
Uno de los principios desarrollados por Charles Darwin constituye la ancestría en común de todos los organismos vivos – es decir, todos compartimos un ancestro remoto.
En “El Origen de las Especies” Darwin plantea la metáfora del “árbol de la vida”. De hecho, usa un árbol gráfico hipotético para desarrollar su idea (curiosamente, es la única ilustración del Origen).
La representación de esta metáfora es lo que conocemos como árboles filogenéticos, que nos permiten exhibir de manera gráfica la historia y las relaciones de algún grupo concreto de organismos.
En los árboles filogenéticos, podemos distinguir las siguientes partes – siguiendo con la analogía botánica:
Ramas: se llama “ramas” a las líneas del árbol, y estas representan a las poblaciones de estudio en el tiempo. Dependiendo del tipo de árbol (ver más adelante), la longitud de la rama puede o no tener un significado.
En la punta de las ramas encontramos a los organismos que queremos evaluar. Estos pueden ser entes que estén vivos actualmente, o bien seres extintos. Las especies serían las hojas de nuestro árbol.
Raíz: la raíz es la rama más ancestral del árbol. Algunos lo tienen y se denominan árboles enraizados, mientras que otros no lo tienen.
Nodos: los puntos de ramificación de las ramas en dos o más linajes se denominan nodos. El punto representa el ancestro en común más reciente de los grupos descendientes (nótese que estos ancestros son hipotéticos).
La existencia de un nodo implica un evento de especiación – creación de nuevas especies. Luego de esto, cada especie sigue su rumbo evolutivo.
Además de estos tres conceptos básicos, existen otros términos necesarios en lo que respecta a los árboles filogenéticos:
Politomia: cuando en un árbol filogenético presenta más de dos ramas en un nodo se dice que existe una politomia. En estos casos, el árbol filogenético no está resuelto totalmente, porque las relaciones entre los organismos implicados no están claras. Esto suele suceder por la falta de datos, y solo puede solucionarse cuando un investigador acumule más.
Grupo externo: en los temas filogenéticos es común escuchar el concepto de grupo externo – también llamado outgroup. Este grupo se selecciona para poder enraizar el árbol. Debe escogerse como un taxón que divergió con anterioridad del grupo de estudio. Por ejemplo, si estoy estudiando equinodermos, puede usar de outgroup a las ascidias.
Existen tres tipos básicos de árboles: los cladogramas, los árboles aditivos y los árboles ultramétricos.
Los cladogramas son los árboles más simples y exhiben la relación de los organismos en términos de ancestría común. La información de este tipo de árbol reside en los patrones de ramificación, ya que el tamaño de las ramas no tiene ningún significado adicional.
El segundo tipo de árbol es el aditivo, también llamados árboles métricos o filogramas. La longitud de las ramas está relacionada con la cantidad de cambio evolutivo.
Finalmente, tenemos a los árboles ultramétricos o dendogramas, donde todas las puntas de los árboles están a la misma distancia (lo que no ocurre en el filograma, donde una punta puede aparecer más abajo o más arriba que su compañera). La longitud de la rama está relacionada con el tiempo evolutivo.
La elección del árbol está relacionada directamente con la pregunta evolutiva que queramos responder. Por ejemplo, si nos importan únicamente las relaciones entre los individuos, un cladograma será suficiente para el estudio.
Aunque los árboles filogenéticos suelen ser gráficos de amplio uso en biología evolutiva (y en la biología general), existen muchos estudiantes y profesionales que malinterpretan el mensaje que estos gráficos – aparentemente simples – pretenden entregar al lector.
El primer error es leerlos de manera lateral, asumiendo que la evolución implica progreso. Si entendemos correctamente el proceso evolutivo, no hay ninguna razón para pensar que a la izquierda se encuentran las especies ancestrales y a la derecha las especies más avanzadas.
Aunque la analogía botánica de árbol es muy útil, llega un punto donde ya no es tan exacta. Existe una estructura crucial del árbol que no está presente en el árbol: el tronco. En los árboles filogenéticos no encontramos ninguna rama principal.
Específicamente, algunas personas podrían considerar al hombre como la “meta” última de la evolución, y por lo tanto la especie Homo sapiens siempre debería ubicarse como ente final.
No obstante, esta visión no es congruente con los principios evolutivos. Si entendemos que los árboles filogenéticos son elementos móviles, podremos situar al Homo en cualquier posición terminal del árbol, ya que esta característica no es relevante en la representación.
Una característica vital que debemos entender de los árboles filogenéticos es que representan gráficos no estáticos.
En ellos, todas estas ramas pueden rotar – de la misma manera que un móvil puede hacerlo. No queremos decir que las ramas las podemos mover a nuestro antojo, porque algunos movimientos implicarían el cambio del patrón o topología del árbol. Lo que podemos rotar son los nodos.
Para interpretar el mensaje de un árbol, no debemos enfocarnos en la punta de las ramas, debemos hacerlo en los puntos de ramificación, que son el aspecto más importante del gráfico.
Además, debemos tener en cuenta que hay diversas maneras para dibujar un árbol. Muchas veces depende del estilo del libro o de la revista y los cambios en la forma y posición de las ramas no afectan a la información que nos quieren transmitir.
Cuando vamos a referirnos a las especies actuales no debemos aplicarles connotaciones de ancestrales. Por ejemplo, cuando pensamos en las relaciones que hay entre los chimpancés y los humanos, podríamos entender erróneamente que los chimpancés son ancestrales con respecto a nuestro linaje.
No obstante, el ancestro en común de los chimpancés y los humanos no era ninguno de los dos. Pensar que el chimpancé es ancestral sería asumir que su evolución se detuvo una vez que ambos linajes se separaron.
Siguiendo la misma lógica de estas ideas, un árbol filogenético tampoco nos indica si hay especies jóvenes. Como las frecuencias alélicas están en cambio constante y hay nuevos caracteres cambiando en el tiempo, es difícil determinar la edad de una especie y, ciertamente, un árbol no nos otorga dicho dato.
El “cambio de las frecuencias alélicas en el tiempo” es la manera en que la genética de poblaciones define a la evolución.
Al observar un árbol filogenético debemos comprender que este gráfico es simplemente una hipótesis generada a partir de una evidencia concreta. Puede que si añadimos más caracteres al árbol, este modifique su topología.
La pericia de los científicos al escoger los mejores caracteres que permitan dilucidar las relaciones de los organismos en cuestión es clave. Además, existen herramientas estadísticas muy poderosas que permiten a los investigadores evaluar los árboles y elegir el más plausible.
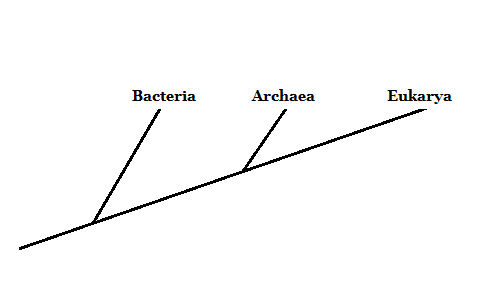
En el año 1977, el investigador Carl Woese propuso agrupar a los organismos vivos en tres dominios: Archaea, Bacteria y Eukarya. Este novedoso sistema de clasificación (anteriormente eran solo dos categorías, Eukaryota y Prokaryota) se basó en el marcador molecular ARN ribosomal.
Las bacterias y los eucariotas son organismos ampliamente conocidos. Las arqueas suelen confundirse con las bacterias. Sin embargo, estas difieren profundamente en la estructura de sus componentes celulares.
Por ello, aunque son organismos microscópicos como las bacterias, los integrantes del dominio Archaea están más relacionados con los eucariotas – porque comparten un ancestro en común más cercano.
Dentro de la biología evolutiva, uno de los temas más controvertidos es la evolución del hombre. Para los oponentes de esta teoría, no tiene lógica una evolución partiendo de un ancestro simiesco que dio origen al hombre actual.
Un concepto clave es entender que no evolucionamos de simios actuales, sino que compartimos un ancestro en común con ellos. En el árbol de los simios y los humanos resalta que lo que conocemos como “simio” no es un grupo monofilético válido, ya que excluye al humano.

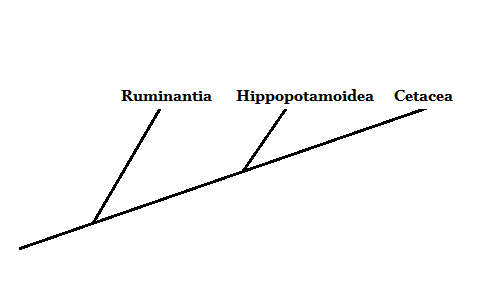
Evolutivamente hablando, los cetáceos representaban un grupo de vertebrados cuyas relaciones con el resto de sus compañeros mamíferos no estaba muy clara. Morfológicamente, las ballenas, delfines y demás miembros tienen pocas semejanzas con el resto de los mamíferos.
Actualmente, gracias al estudio de distintos caracteres morfológicos y moleculares se ha podido concluir que el grupo hermano de los grandes cetáceos está formado por los artiodáctilos – ungulados con pezuñas pares.
- Baum, D. A., Smith, S. D., & Donovan, S. S. (2005). The tree-thinking challenge. Science, 310(5750), 979-980.
- Curtis, H., & Barnes, N. S. (1994). Invitation to biology. Macmillan.
- Hall, B. K. (Ed.). (2012). Homology: The hierarchical basis of comparative biology. Academic Press.
- Hickman, C. P., Roberts, L. S., Larson, A., Ober, W. C., & Garrison, C. (2001). Integrated principles of zoology. McGraw–Hill.
- Kardong, K. V. (2006). Vertebrates: comparative anatomy, function, evolution. McGraw-Hill.
- Kliman, R. M. (2016). Encyclopedia of Evolutionary Biology. Academic Press.
- Losos, J. B. (2013). The Princeton guide to evolution. Princeton University Press.
- Page, R. D., & Holmes, E. C. (2009). Molecular evolution: a phylogenetic approach. John Wiley & Sons.
- Rice, S. A. (2009). Encyclopedia of evolution. Infobase Publishing.
- Starr, C., Evers, C., & Starr, L. (2010). Biology: concepts and applications without physiology. Cengage Learning.